

2021年11月1日,浙江大学第二医院医学遗传科/罕见病诊疗中心成立。吴志英(前排中)、于浩(后排左二)等人合影。受访者供图
浙江大学医学院附属第二医院(以下简称浙大二院)7号楼6楼,U型走廊里有12个病房,住着23个不同种类的罕见病患者。他们中的大多数人不能像正常人一样使用他们的腿。病情轻的可以蹒跚前行,病情重的需要轮椅。其中,还有不少患者手指颤抖,说话含糊不清。
这里是浙大二院医学遗传科/罕见病诊疗中心。前身为神经内科第五病房,成立于2021年11月1日。是国内唯一的罕见病病房。这里床位紧张,只要有人出院,马上就会有人入住。一面墙配有扶手,专门配有肌肉活检室和肌电图室。
55岁的浙大二院医学遗传科主任吴志英教授是这个病房的发起者和负责人。吴志英经历了罕见病逐渐被看到的过程,也见证了一些罕见病治疗药物进入医保后对患者的影响。吴志英说,对于这些医务人员来说,在罕见病领域没有什么特别吸引眼球的故事,“因为这就是每天发生在我们身边的事情。我们希望每个人都能以平常心对待罕见疾病,只有这样才能真正公平对待这些患者。”
患者
25岁的杭州人刘墨于2月23日住进该病房。他患有脊髓性肌萎缩症(SMA),下肢肌肉萎缩,无法自由活动。做检查的时候,他需要用有些无力的手撑着床,然后靠着父亲的力量把全身转移到轮椅上。SMA是一种罕见的遗传性神经肌肉疾病。我国新生儿SMA发病率约为1/6000 ~ 1/10000。目前全国约有3万患者。
刘墨在12岁时被确诊。“那时候中国基本没有医生和药品。去外省看病了。医生说我是全院唯一一例。”没有别的办法,刘墨能做的只有等待。2016年,诺西奈德钠在美国获得批准,2019年获得美国食品药品监督管理局批准,成为中国首个治疗脊髓性肌萎缩症的药物。2021年12月3日进入调整后的国家医保药品目录,每针从70万元降至3.3万元。从生病到使用这种救命药,刘墨等了13年。
东东和刘墨住在一个病房。东东在小学三年级。他有着圆圆的鼻子和鸡蛋般的皮肤,一双眼睛,一身深绿色的睡衣和由他母亲缝制的拖鞋。除了用父亲的手机看动画,其他时间他都不肯老老实实躺在床上。无论是起床还是走路,东东都不能像正常孩子一样。他要把整个上半身贴在床上,以肚子为中心把腿移到床上,然后慢慢伸到床下,走路时保持右肩向上拱起。
2016年,东东被确诊为杜氏肌营养不良症,这也是一种罕见的遗传性疾病,主要发生在男性,女性多为无症状携带者。
东东的父亲陈驰说,他带着孩子去过大大小小的医院,这次来浙大二院看看有没有合适的药物和治疗方法。

2022年2月24日,东东在病床上玩手机。新京报记者李摄
2月24日,上午10点,住在东东隔壁房间的安然换了病号服。4点开始准备出院,在病房待了一个星期。他感叹自己“终于可以出去了”。这是他第七次住院,已经很熟悉了。他的疾病“肝豆状核变性”很少给他带来任何心理困扰。
2月18日,安然住在房子里,因为它在过年期间摄入了大量含铜量高的山核桃,导致症状加重。正常人可以通过尿液和汗液将人体内多余的铜排出体外,但安然的一个基因存在缺陷,使得他体内的铜无法顺利排出体外。
安然第一次发病是在将近十年前。职校毕业后做电工,手抖得厉害,口水直流。
随后的几年,他被父母领着去了各大医院。起初,他被误诊为双相情感障碍,吃了很多精神类药物,“但没有效果。”直到2020年9月,在浙大二院做基因检测后,被确诊为肝豆状核变性。此时,他已经与这种奇怪的、闻所未闻的疾病生活了近十年。
肝豆状核变性作为一种罕见病,于2018年被列入国家卫健委等五部门联合发布的首批罕见病目录。它是一种遗传性铜代谢障碍,累及脑和肝,发病率约为万分之一至三万分之一。
安然没听说过“中国唯一的罕见病病房”,但他看病多年,确实没见过一个病房有这么多罕见病患者。“大部分都和我一样,不能走路,手脚发抖。”他热衷于在患者之间穿梭,哪怕不是同一种病,他总能给几句安慰,他的段子脱口而出,“为什么有东京,有南京,有北京,却没有?因为西方经典被唐僧拿走了。”
安然·曹金是病房里为数不多的病人之一。他们年龄差不多,会一起讨论游戏和暗恋的女生。如果没有必要,曹金不会下床并在医院里呆四天。曹金在病房的U形走廊里走了不到十次。他害怕安静的走廊里其他病人及其家属的目光。"他们的眼神似乎在说他们瞧不起我。"他宁愿藏在人群深处。住院让他的生活更有规律。当他每天早上醒来时,他无事可做。他拿出手机,看了一小段视频,又按了一下。时间随着输液瓶里的生理盐水流逝,他又睡着了。
曹金于2020年9月被诊断患有脊髓小脑性共济失调,这是一种常染色体显性遗传病。如果父母一方是患者,有一半的概率会遗传给下一代。他的母亲在2018年死于同样的疾病。她去世前坐了十多年轮椅。曹金担心他会有同样的命运。他坚持跑步,希望身体机能的衰退晚一点到来。
隔离病房
吴志英第一次接触罕见疾病是在30年前。那时候还没有罕见病的名称。“我们通常称之为遗传病。”1992年,吴志英师从福建省神经病学奠基人之一慕容沈星教授,获得神经病学硕士学位。有一天,门诊里,来了一个坐轮椅的少年。他身体扭曲,四肢僵硬,口齿不清,口水不停地流。“我的导师用手电筒照了照他的眼睛,指着角膜缘上一个棕绿色的环说,‘这是角膜K-F环,这个病人是肝豆状核变性病人’。”这是吴志英接触到的第一例肝豆状核变性病例。她被快速诊断出病人的导师高超的医术震惊了。“也希望以后能看到别人看不到的疑难杂症”。就这样,吴志英将他的硕士和博士研究重点放在了肝豆状核变性上,并从肝豆状核变性扩展到了其他罕见疾病。

吴志英回忆说,起初,罕见病的研究面临很多困难,病例收集困难,往往导致诊断瓶颈。“十几年前有个病人来福建看我。起初我无法诊断他,直到十几年后才发现这个病的致病基因。确诊前我给他做了基因检测,叫他回来复诊。”
吴志英的医学生涯从福建到上海,又从上海到杭州。多年来,她在电脑里保存了3万多份罕见病患者的病历,但并不是每个患者都能在门诊确诊。吴志英说,病人通常在10年前就康复了。“由于技术原因,10多年前我们无法诊断一些罕见疾病。随着科技的发展和经验的提高,我们的诊断能力也有了很大的提高。如果从最前沿的新闻中确定患者患的是哪种罕见病,我们会拿出病历,通过各种方式联系患者。”在接触患者的时候,我们会遇到各种各样的困难。有些病人只留下不再使用的呼机号码。吴志英团队只能写信到开头留下的地址。如果联系不上他们,队里就给村委会打电话。

2022年2月24日,吴志英在病房查房。新京报记者李摄
在吴志英看来,基因测序等基因诊断技术在临床上的广泛应用,可以视为罕见病诊疗的分水岭。在此之前,罕见病主要通过临床表现和常规实验室检查方法进行判断,无法在分子水平上进一步诊断疾病。基因检测被认为是诊断大多数罕见疾病的金标准。吴志英说,在中国,近10年来,基因检测被广泛用于诊断罕见病和遗传病。人类基因组有两万多个基因,致病基因的发现还在不断更新。
吴志英酝酿了很久,准备设立一个罕见病病房。“把病房分开,让罕见病患者找地方,医生看病,可以为罕见病患者提供更有针对性的治疗,也有助于我们对罕见病患者进行随访”。
2021年11月1日,浙大二院在国内首次成立医学遗传科/罕见病诊疗中心,成为一个独立的临床科室,设有病房、门诊和医学遗传实验室,专治罕见病。采用病房、门诊、实验室三位一体模式,精准诊治罕见病。
见过的罕见病
能留在病房的,都是不幸命运中的幸运儿。他们可以在病房里进行康复治疗,延缓疾病对身体的危害。
资料显示,目前全世界已知的罕见病有7000多种,但其中只有6%左右可以用药物治疗。更糟糕的是,80%的罕见病都是有遗传问题的遗传病。
我国一半以上的罕见病患者面临着海外有药国内无药的情况,部分家庭不得不从海外高价购买。在我国发布的121种罕见病中,有74种对应的罕见病治疗药物在国内外上市销售,涉及100多种药物。而我国只有31种罕见病有治疗药物,涉及50多种药物。
如今,身为中国罕见病联盟浙江合作组主席的吴志英回忆起,中国对罕见病诊疗的广泛关注始于2018年,当时国家五部委发布了《罕见病目录》,将121种疾病列入罕见病名单,其中包括安然的Wilson's病和曹金的脊髓小脑性共济失调。2019年,国家卫健委宣布建立全国罕见病诊疗协作网,在全国范围内选择324家罕见病诊疗能力强、病例量大的医院组成罕见病诊疗协作网。这些国家层面的行动,让罕见病患者从隐形到被更多人看到。
在吴志英看来,目前我国罕见病的诊断水平已经可以与国际接轨,但在治疗方面,直到现在,还没有原创的罕见病治疗药物。目前国内大量的药品要么是从国外进口的,要么是仿制的,导致中国市场上罕见病的药品极其昂贵。“我们非常希望未来能有更多关于罕见病治疗的研究,国家能投入更多资金,社会和企业也能给予更多支持,研究罕见病的治疗靶点和药物。如果有足够的资助,我相信中国科学家可以研究罕见病的发病机制,找到相关靶点,开发原创药物。”

2022年2月21日,吴志英门诊。新京报记者李摄
2021年12月,一段医保局“灵魂砍价”的视频火遍网络,罕见病用于医保的新闻被更多人关注。吴志英明显感觉到来门诊的人多了,回来看病的人也多了。“有的人病了十几年,主要是我们没有药,药价贵。即使确诊了,病人因为经济问题来了这么一次就再也不会来了。”
截至2021年12月,我国共有60余种罕见病药品获批上市,其中涉及25种罕见病的40余种药品纳入国家医保目录。如果算上第一批罕见病目录中的121种疾病,大概有20%可以纳入医保,比如肝豆状核变性的青霉胺、脊髓性肌萎缩症的去甲肾上腺素钠、法布里氏病的琼脂酶α等罕见病,都在医保范围内。但病房主治医生余浩解释说,只统计罕见病的药物来计算有多少罕见病患者被医保覆盖是不科学的。“有很多药物不是针对罕见病的,而是常用于常见病,而且常用药物相对便宜。还能减轻患者的经济负担。”
从贵药到平民药
在浙大二院的罕见病病房里,有些病人是为了诊断而住院,有些是为了康复。虽然很多罕见病没有特效药,但是固定的康复治疗可以缓解患者的症状。安然说,他来医院之前手脚发抖,按时在医院吃药,出院后病情稳定。
这次刘墨住院是为了注射院内钠,这是国内第一个批准用于治疗脊髓性肌萎缩症的进口药。上市之初被称为高价药,每针70万元。
刘墨身体状况最差的时候,曾经想过直接注射,但最后被价格吓退了。
病房主治医生余浩说,“院内钠的注射,前两个月需要四针,之后每四个月一针,第一年倒数六针,之后每年三针,不管大人小孩,目前的方案都一样”。
刘墨的账,从2019年院内钠在国内上市开始算。一针70万,第一年420万,以后每年210万,住院费用每年1万。没有药物治疗,开始后的每一年都是“无尽的支出”,他不敢想。他真正第一次打针的想法,是当年SMA协会组织的一个援助项目。“大概是55万打针,买一送五,第二年送两个”。虽然价格还是偏高,但是“少部分患者能接受。”刘墨的父母也鼓励他去尝试。“如果不打针,病情会很快恶化,他们认为会持续一两年。”但他犹豫了一下,然后放弃了,一方面是经济原因,另一方面是自己“病情稳定,还没到着急的时候”。
2021年12月3日,国家医疗保障局公布了最新版本的国家医保药品目录调整结果。诺西奈德钠注射液进入医保药品目录,费用从近70万元/针降至3.3万元/针,将于2022年1月1日实施。根据各地医保覆盖比例不同,患者需要自费的费用从每针1000元到20000元不等。
刘墨一听到药品降价的消息,就在浙大二院预约了去甲酸钠的治疗。2月24日,他进行了第一次注射。
余浩对药物的疗效有信心。"注射后,患者会感觉更强壮,如行走和站立更容易."
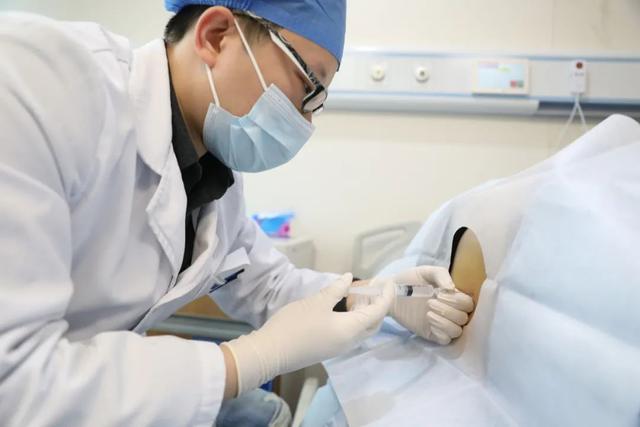
2022年1月1日,于浩给患者注射了诺西奈德钠注射液。受访者供图
东东的父亲陈驰对刘墨表示羡慕。“人们有治疗这种疾病的药,我们还不知道什么时候能战胜它。”
余浩说,目前国内没有针对杜氏肌营养不良症的特效药,国外上市的基因治疗药物也很少。然而,这些药物价格昂贵,20kg儿童每年的治疗费用超过300万人民币。显然,陈驰负担不起这样的治疗费用。
陈驰还是带着儿子去了医院,心想:“如果你能留在医院,下次有药,医生肯定会第一个联系我们。”
患病十年,安然在肝豆状核变性的治疗上花了十几万。青霉胺,一种用于治疗的药物,已经纳入医保,农村医保,杭州西湖易联宝可以在一定程度上缓解安然的经济压力。
于浩说,“去年进入医保目录的7种药品,为罕见病患者减轻了不少负担,尤其是降纤酸钠。下降幅度如此之大,以至于接受治疗的患者数量明显增加。去年我们科室只收治了3名患者,但从年初到现在,我们已经注射了13名患者。”
刘墨在医院注射了第一针钠后已经出院,他感到幸运的是,他可以等待一种救命的药物。“慢慢会好起来的,就一点点希望。”
(本文患者及家属均为化名)
新京报记者李编辑校对吴兴发







